A visual explanation of decision trees with a simple example.
Category Archives: Machine Learning
Learning to learn – supervised versus unsupervised machine learning
In this blog post, I would like to introduce the two main forms of machine learning, supervised and unsupervised machine learning. The two differ quite a lot in the task they address, in the data that is necessary and in the algorithms that are used.
Supervised learning starts out from a set of data where each item is associated with a label that indicates a category. One example data set could be a collection of e-mails where each one is labeled as “spam” or “non-spam“. Another example data set could be a photo collection with categories such as “shows a mountain“, “is a portrait” or “taken at night“. These labels have usually been assigned by a human. The task for the machine learning algorithm is now to learn how to assign these labels. To this end, it is shown a large number of items with labels and it tries to learn how to distinguish one category from the other. The process is similar to a human who tries to learn something new. A child might first call everything with four legs a cat, but after seeing enough animals and the accompanying comment “no, that’s not a cat, that’s a X“, she will over time come to distinguish actual cats from dogs, cows or horses. Supervised machine learning algorithms do basically the same thing. Given a large amount of examples and their category, they try to find features that separate one class from the others. Coming back to the example of e-mails, the algorithm may find that e-mails that contain the phrases “earn a lot of money” or “prince from Nigeria” are likely spam. Or in the case of photos, it may learn that when a picture is dark, it has been taken at night. There are two main differences to the learning process of us humans. One disadvantage is, that the algorithm cannot generalize as well as we do. But this is offset by the advantage that it is much faster than we are and can look at a much larger data set than we ever could. Supervised learning is sometimes also called classification and there are many machine learning algorithms available. Examples include decision trees, Naive Bayes, logistic regression and neural networks.
Let us now turn to unsupervised learning. Just like with supervised learning, we start with a large data set to show the computer. But in contrast to supervised learning, there are no labels. No one is telling the algorithm what to learn. The task is rather to use the internal characteristics of the data to come up with groups inside the data. For example we could try to find groups of users with similar shopping habits out of all the online customers of your company. Or products that are similar to each other in the set of items those sold at a web shop. Or group the web pages in the result of a web search, e.g., the pages discussing jaguar the car versus those about the cat. The resulting division in the data is not based on outside input, like it is for classification, where a human has to define the categories for the data beforehand. The division is only based on the similarity of items in the data set among each other. No human has defined that for the search “jaguar” there are results for a cat and a car, but just by looking at the pages it turns out that there are two groups of pages that use a very different vocabulary. Algorithms for unsupervised learning include clustering algorithms and methods for covariance analysis like principal component analysis/singular value decomposition.
For the sake of completeness, let me mention that supervised and unsupervised learning are the two poles of machine learning methods, but not everything falls clearly into one camp or the other. Several semi-supervised approaches exist that fall somewhere in between. Some of these approaches use partial labels or external information to create the data set from where supervised learning can then start. Other methods use supervised learning to incrementally increase the data set on which the learning algorithm itself is trained. And of course there is no limit to creativity in this area.
To summarize, supervised and unsupervised learning differ in the task they want to solve (supervised learning assigns human-defined categories while unsupervised learning tries to find inherent groups in the data), the data that is necessary (supervised learning needs a set of items with associated categories, unsupervised learning needs only the items) and in the algorithms that are used (classification algorithms for supervised learning versus clustering algorithms for unsupervised learning).
This post has first appeared at 5analytics.com
Learning to learn – What to look for in the evaluation of classification
Applying some machine learning algorithm to classify some data is made easy these days. There is a large amount of programming libraries, applications and online services available on the market. But how do we know whether the algorithm works? Or which of the methods to chose from is the best for our task? This post will give a very short introduction to the four most relevant issues in the evaluation of classification results. None of these points is restricted to a specific machine learning algorithm. In fact, none of them requires any understanding of the used classification method at all.
1. Good machine learning requires good data
The first and most obvious point is, that machine learning requires training data. Training data consists of a number of data items with associated labels assigned by humans. For example, we could train on a set of e-mails which have been labeled as spam or non-spam by the person receiving them. In order for a learning algorithm to learn something useful, a few conditions should be met by the training data. First, there should be enough data. Learning from only 100 e-mails will not work, as there are many types of spam mails. Second, the data should be as clean as possible. If half of the spam-labels are wrong, there is no way how an algorithm can learn what real spam is. And finally, the data should also be as close as possible to the real data that you want to classify later. Training a spam-recognition system on English and then apply it to German will probably not work well.
2. Don’t evaluate on your training data
After the machine learning algorithm has learned to distinguish the classes on the training data, it is ready to be applied to new data. Based on what it has learned from the training data, the algorithm will assign a class to each new data item. A common beginner’s mistake is to apply the algorithm to the training data again. This will lead to very good results, but these results are misleading. Imagine that the “learning algorithm” is just memorizing complete e-mails. If the exact same e-mail is shown to the algorithm again, it will confidently assign the correct class. 100% of training set e-mails will be correct! But even changing one word will cause the algorithm to fail, so it is no use to us in reality. Of course real learning algorithms are more complex, but the issue is the same. It is very easy to be confident about what you already have seen. The hard part is to deal with new stuff. So in order to have a reliable evaluation, the algorithm should be trained on one data set and applied to another totally separate set.
3. Evaluate on data that is close to the data you want to classify later
As we have just discussed, we need to evaluate on data that is different from the training data. But, just like the training data, the evaluation data should be as close as possible to the real data that you want to classify later. If you want to classify German, it doesn’t help you to know that the spam-classifier works very well on English. A common procedure for a good evaluation is to create one data set with labeled data, and then split it up into training and test data (e.g., 80% training data, 20% test data). No item is allowed to be in both sets at the same time. Another common technique is called k-fold cross-validation. This method splits the data into k (often 10) folds and does k train-test runs. In each run, one of the folds is used as test data and the other folds are used as training data. The folds do not change between runs, so in the end every item in the data has been assigned a label, but at that point this item was not in the training set, so point 2 is not violated. For both technique it is worth thinking about whether to randomly shuffle the folds or to enforce a similar label-distribution in all the folds in order to avoid artificial inflation of the results.
4. Chose the right evaluation metric for your problem
After the machine learning system has assigned a class to every data item, we compare the assigned labels to the real labels. The larger the percentage of correct labels, the better the system. There are many ways of comparing the labels depending on the nature of the labels and their distribution. The simplest measure, called accuracy, is to count the number of correct assignments, e.g., how many real spam-mails have been classified as spam by the system and how many non-spam-mails have been classified as non-spam. But accuracy is not a good measure in some cases. Let’s assume that 90% of mails are non-spam. If a system always assigns the label non-spam, it will be 90% accurate – but not useful at all. The same thing happens with many classes if some are much bigger than the others. Accuracy is also not a good choice when labels are on a scale. In this case confusing 1 and 5 is much more serious than confusing 1 and 2 and accuracy does not reflect this. There are alternative metrics for such scenarios that should be used.
I will stop here, although there is more much to be said. I encourage everybody to investigate the topic in more detail. Good evaluation is at least as important as good machine learning algorithms. If evaluation numbers do not reflect the expected real performance of a system, how can they be the basis of any decision?
This post has first appeared at 5analytics.com
Precision, Recall and F-measure
In the last post we discussed accuracy, a straightforward method of calculating the performance of a classification system. Using accuracy is fine when the classes are of equal size, but this is often not the case in real world tasks. In such cases the very large number of true negatives outweighs the number of true positives in the evaluation so that accuracy will always be artificially high.
Luckily there are performance measures that ignore the number of true negatives. Two frequently used measures are precision and recall. Precision P indicates how many of the items that we have identified as positives are really positives. In other words, how precise have we been in our identification. How many of those that we think are X, really are X. Formally, this means that we divide the number of true positives by the number of all identified positives (true and false):
")
Recall R indicates how many of the real positives we have found. So from all of the positive items that are there, how many did we manage to identify. In other words, how exhaustive we were. Formally, this means that we divide the number of true positives by the number of all existing positives (true positives and false negatives):
")
For our example from the last post, precision and recall are as follows:
 = 1/4 = 0.25")
 = 1/3 = 0.33")
It is easy to get a recall of 100%. We just say for everything that it is a positive. But as this will probably not the case (or else we have a really easy dataset to classify!), this approach will give us a really low precision. On the other hand, we can usually get a high precision if we only classify as positive one single item that we are really, really sure about. But if we do that, recall will be low, as there will be more than one item in the dataset to be classified (or else it is not a very meaningful set).
So recall and precision are in a sort of balance. The F1 score or F1 measure is a way of putting the two of them together to produce one single number. Formally it calculates the harmonic mean of the two numbers and weights the two of them with the same importance (there are other variants that put more importance on one of them):
/(P + R)")
Using the values for precision and recall for our example, F1 is:
/(0.25 + 0.33) = 0.165 / 0.58 = 0.28")
Intuitively, F1 is between the two values of precision and recall, but closer to the lower of the two. In other words, it penalizes if we concentrate only on one of the values and rewards systems where precision and recall are closer together.
Link for a second explanation: Explanation from an Information Retrieval perspective
Accuracy
We are still trying to figure out how good our system for determining whether e-mails are spam or not is. In the last post we ended up with a confusion matrix like this:
| Actual label | |||
| Spam | NonSpam | ||
| Predicted label | Spam | 1 (true positives, TP) | 3 (false positives, FP) |
| NonSpam | 2 (false negatives, FN) | 4 (true negatives, TN) | |
Now we want to calculate numbers from this table to describe the performance of our system. One easy way of doing this is to use accuracy A. Accuracy basically describes which percentage of decisions we got right. So we would take the diagonal entries in the matrix (the true positives and true negatives) and divide by the total number of entries. Formally:
/(TP+TN+FP+FN)")
In our example the accuracy is:
/(1+4+2+3) = 5/10 = 0.5")
Using accuracy is fine in examples like the above when both classes occur more or less with the same frequency. But frequently the number of true negatives is larger than the number of true positives by many orders of magnitudes. So let’s assume 994 for true negatives and when we calculate accuracy again, we get this:
/(1+994+2+3) = 995/1000 = 0.995")
It doesn’t really matter if we correctly identify any spam mails. Even if we always say NonSpam, so we get zero Spam-Mails right, we still get more nearly the same accuracy as above. So accuracy is not a good indicator of performance for our system in this situation. In the next post we will look at other measures we can use instead.
Link for a second explanation: Explanation from an Information Retrieval perspective
Confusion matrix
Let’s say we want to analyze e-mails to determine whether they are spam or not. We have a set of mails and for each of them we have a label that says either "Spam" or "NotSpam" (for example we could get these labels from users who mark mails as spam). On this set of documents (the training data) we can train a machine learning system which given an e-mail can predict the label. So now we want to know how the system that we have trained is performing, whether it really recognizes spam or not.
So how can we find out? We take another set of mails that have been marked as "Spam" or "NotSpam" (the test data), apply our machine learning system and get predicted labels for these documents. So we end up with a list like this:
| Actual label | Predicted label | |
|---|---|---|
| Mail 1 | Spam | NonSpam |
| Mail 2 | NonSpam | NonSpam |
| Mail 3 | NonSpam | NonSpam |
| Mail 4 | Spam | Spam |
| Mail 5 | NonSpam | NonSpam |
| Mail 6 | NonSpam | NonSpam |
| Mail 7 | Spam | NonSpam |
| Mail 8 | NonSpam | Spam |
| Mail 9 | NonSpam | Spam |
| Mail 10 | NonSpam | Spam |
We can now compare the predicted labels from our system to the actual labels to find out how many of them we got right. When we have two classes, there are four possible outcomes for the comparison of a predicted label and an actual label. We could have predicted "Spam" and the actual label is also "Spam". Or we predicted "NonSpam" and the label is actually "NonSpam". In both of these cases we were right, so these are the true predictions. But, we could also have predicted "Spam" when the actual label is "NonSpam". Or "NonSpam" when we should have predicted "Spam". So these are the false predictions, the cases where we have been wrong. Let’s assume that we are interested in how well we can predict "Spam". Every mail for which we have predicted the class "Spam" is a positive prediction, a prediction for the class we are interested in. Every mail where we have predicted "NonSpam" is a negative prediction, a prediction of not the class we are interested in. So we can summarize the possible outcomes and their names in this table:
| Actual label | |||
| Spam | NonSpam | ||
| Predicted label | Spam | true positives (TP) | false positives (FP) |
| NonSpam | false negatives (FN) | true negatives (TN) | |
The true positives are the mails where we have predicted "Spam", the class we are interested in, so it is a positive prediction, and the actual label was also "Spam", so the prediction was true. The false positives are the mails where we have predicted "Spam" (a positive prediction), but the actual label is "NonSpam", so the prediction is false. Correspondingly the false negatives, the mails we should have labeled as "Spam" but didn’t. And the true negatives that we correctly recognized as "NonSpam". This matrix is called a confusion matrix.
Let’s create the confusion matrix for the table with the ten mails that we classified above. Mail 1 is "Spam", but we predicted "NonSpam", so this is a false negative. Mail 2 is "NonSpam" and we predicted "NonSpam", so this is a true negative. And so on. We end up with this table:
| Actual label | |||
| Spam | NonSpam | ||
| Predicted label | Spam | 1 | 3 |
| NonSpam | 2 | 4 | |
In the next post we will take a loo at how we can calculate performance measures from this table.
Link for a second explanation: Explanation from an Information Retrieval perspective
Euclidean and cosine distance for unit vectors (and negative entries!)
Just a few quick words about the assumption we made in the last post about all our entries in the vectors being positive so that we can define the cosine distance as 1 minus the similarity. This assumption is actually not necessary. We can have negative entries, as long as our vectors are normalized to unit length everything still works.
Remember Euclidean distance for unit vectors:
 = \sqrt{2(1 - \sum_i p_i q_i)}")
And cosine similarity for two unit vectors:
 = \sum_i p_i q_i")
So now, like we did in the last post, let’s say we have two vectors v and w and we know that measured with Euclidean distance, v is closer to some other point p than w*:
 \leq d_{\text{euclid}}(\vec{p},\vec{w})")
We do the same steps as in the last post, but then go on and get rid of the 1 and the minus (attention, this changes the direction of the inequality):



Voila, cosine similarity!
So if p is closer to v than to w as measured with Euclidean distance, the cosine similarity of p and v is higher than that of p and w:
 \leq d_{\text{euclid}}(\vec{p},\vec{w}) \Leftrightarrow s_{\text{cosine}}(\vec{p},\vec{v}) \geq s_{\text{cosine}}(\vec{p},\vec{w})")
So whenever you have unit length vectors and are only interested in relative distances, it shouldn’t make a distance whether you use Euclidean distance or cosine similarity.
* Same footnote as last time: The text says “closer” and not “closer or the same” and that is actually what I wanted to say, but there seems to be some strange bug in this LaTeX plugin that doesn’t allow you to use the < sign in a formula... so we'll take the less-or-equal sign and just ignore the equal-part.
Euclidean and cosine distance for unit vectors
The Euclidean distance between two vectors p and q is the length of the line segment that connects them (here and in all following formulas the sum is over all dimensions of the vectors, i.e., if we have n dimensions the sum ranges from i=0 to n):
 = |\vec{p} - \vec{q}| = \sqrt{\sum_i (p_i - q_i)^2}")
Using the binomial expansion, we can write this as follows:
 = \sqrt{\sum_i p_i^2 - 2\sum_i p_i q_i +\sum_i q_i^2}")
Unit vectors have a length of 1 (by definition), length is calculated as the Euclidean norm, that is, the Euclidean distance of a vector to the zero vector, i.e., the square root of the sum of all sqared entries in the vector:
 = \sqrt{\sum_i (p_i-0)^2 } = \sqrt{\sum_i p_i^2 }")
If something is 1, its square is also 1:

We can now replace the squared sums over all vector elements in the formula for Euclidean distance with 1:
 = \sqrt{1 - 2\sum_i p_i q_i + 1} = \sqrt{2 - 2\sum_i p_i q_i} = \sqrt{2(1 - \sum_i p_i q_i)}")
Now let’s see how the cosine distance is defined. The more common thing to do is to calculate the cosine similarity of two vectors as the cosine of the angle between them:
 = \frac{\vec{p} \cdot \vec{q}}{|\vec{p}| |\vec{q}|} = \frac{\sum_i p_i q_i}{|\vec{p}| |\vec{q}|}")
As we have unit vectors, we can get rid of the division by the length (which is always 1), so the formula is simplified to the dot product between the two vectors:
When we have a vector space where the entries correspond to occurrences of terms in a document, all entries are positive, so the value of the cosine similarity will always be between zero and one. This means, we can define the cosine distance as:
 = 1 - s_{\text{cosine}}(\vec{p},\vec{q}) = 1 - \sum_i p_i q_i")
So let’s put it together. Let’s say we have two vectors v and w and we know that measured with Euclidean distance, v is closer to some other point p than w is*:
We can now replace the Euclidean distance with the formula from above, square both sides (because that doesn’t change the inequality relation) and get rid of the two that appears on both sides:
} \leq \sqrt{2(1 - \sum_i p_i w_i)}")
 \leq 2(1 - \sum_i p_i w_i)")

What we are left with is the cosine distance! So, putting start and end together, what we have shown is:
 \leq d_{\text{euclid}}(\vec{p},\vec{w}) \Leftrightarrow d_{\text{cosine}}(\vec{p},\vec{v}) \leq d_{\text{cosine}}(\vec{p},\vec{w})")
This doesn’t mean that when you calculate Euclidean distance and cosine distance between two vectors that you will get the same number. But whenever you are only interested in relative distances (that means you only want to know which of two vectors is closer to something than the other) and you have vectors that are normalized to unit length with only positive entries, then the result should be the same whether you use cosine or Euclidean distance.
* The text says “closer” and not “closer or the same” and that is actually what I wanted to say, but there seems to be some strange bug in this LaTeX plugin that doesn’t allow you to use the < sign in a formula... so we'll take the less-or-equal sign and just ignore the equal-part.
Precision-Recall-Curves and Mean Average Precision
Precision-recall curves are often used to evaluate ranked results of an information retrieval system (e.g., a search engine). The principle is easy, for every search result, check the precision and recall you have until now (precision/recall at k). If you plot this in a graph with recall on the x-axis and precision on the y-axis, you end up with something like this (blue line):
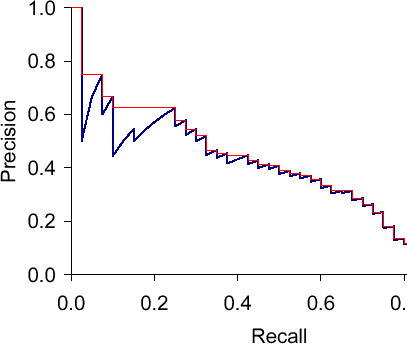
The essential shape is always the same. Why? Let’s say we have looked at k results which corresponds to a point with a precision and a recall value. What can happen when we go to result k+1? The result can be correct, then recall will increase and precision as well – the curve goes up and right. Or it can go wrong, then recall stays the same and precision drops – the curve goes straight down.
The red line is the interpolated precision, meaning we define precision at some arbitrary level to be the maximum precision reached at any later recall level. Essentially, we flatten the "teeth" of the curve. The difference can be pretty big (see in the plot at recall around 0.2), we can even "skip" a tooth.
What would the curve look like for a perfect system? Meaning a system that only returns correct results? It would be 1.0 for every recall level. A system that never returns a correct result? 0 for every recall level.
What should the value be for precision at recall 0? If we interpolate, the answer is clear: the highest precision value at some later recall level. This does not have to be 1.0 – it could happen that the first result is wrong, the second correct, then we have P=0.5 at k=2 and it might only drop from there.
Sancho McCann. It’s a bird… it’s a plane… it depends on your classifier threshold. 2011.
Christopher D. Manning, Prabhakar Raghavan and Hinrich Schütze. Introduction to Information Retrieval. Cambridge University Press, 2008. (Chapter 8)